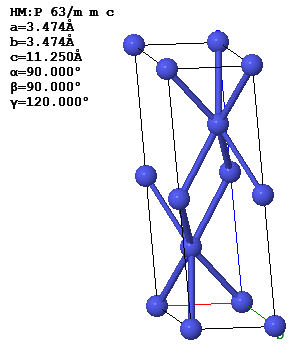
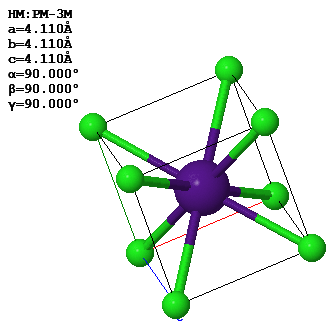
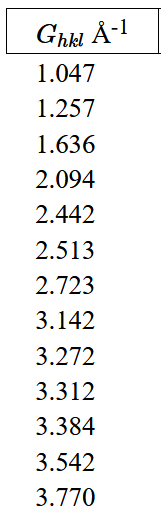Every periodic function has a Bravais lattice and a corresponding reciprocal lattice. The relations between the primitive lattice vectors in real space and in reciprocal space are,
Here $f_{\vec{G}}$ are complex coefficients called the structure factors.
For the exam you should be able to determine the reciprocal lattice vectors of any Bravais lattice and know that the reciprocal lattice of an orthorhombic lattice with lattice constants $(a,b,c)$ is also an orthorhombic lattice $(2\pi /a,2\pi /b,2\pi /c)$ and the reciprocal lattice of fcc is bcc and the reciprocal lattice of bcc is fcc.
Reciprocal space can be divided into Brillouin zones. The first Brillouin zone is the set of points closer to the origin in reciprocal space than to any other reciprocal lattice vector. It is analogous to the Wigner-Seitz cell in real space. All points outside the first Brillouin zone can be reached by a vector in the first Brillouin zone plus a reciprocal lattice vector. You should be able to construct the first Brillouin zone of any reciprocal lattice.
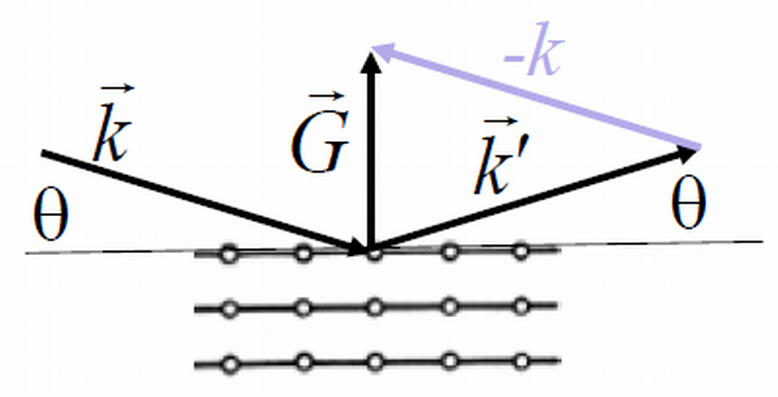
Diffraction occurs when waves strike a periodic structure and the wavelength of the waves is shorter than the periodicity of the structure. Under these conditions, some of the waves will continue in the direction $\vec{k}$ of the primary beam and some will be scattered elastically to directions $\vec{k}'$ where the scattering vector is a reciprocal lattice vector,
This is called the diffraction condition and it is often used to determine crystal structures. In the experiment, a crystal is put in the primary beam of an x-ray diffractometer. For elastic scattering $|\vec{k}|=|\vec{k}'|=2\pi /\lambda$. By detecting the angles at which diffraction peaks are observed, it is possible to calculate the reciprocal lattice vectors from the diffraction condition. If many reciprocal lattice vectors are determined, it is possible to deduce the primitive lattice vectors in reciprocal space,
From the primitive lattice vectors in reciprocal space, it is possible to calculate the primitive lattice vectors in real space (the formulas are given at the top of this page). The primitive lattice vectors in real space determine the Bravais lattice and the volume of the unit cell in real space. The atoms in the basis can be determined by comparing the intensities of the diffraction peaks to the structure factors that appear in the Fourier series for the electron density,
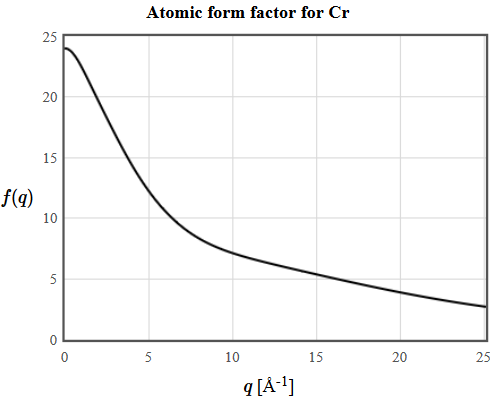
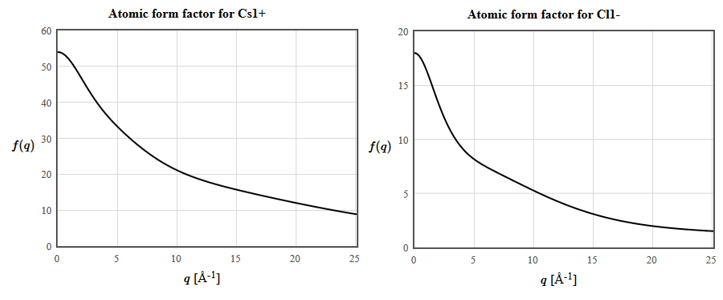
Here $\vec{T}$ are the translation vectors of the Bravais lattice, $n_j\left(\vec{r}\right)$ is the electron density of atom $j$ and $n_{\vec{G}}$ are the structure factors. The intensity of the diffraction peak $\vec{G}$ is proportional to $|n_{\vec{G}}|^2$. Since most electrons are core electrons, the electron density in a crystal is sharply peaked around the nuclei. A simple approximation for the electron density of a unit cell of a crystal is,
To determine how the atoms are arranged in the basis, typically you have to guess the arrangement of the atoms, calculate the resulting structure factors, and them compare them to the intensities of the diffraction peaks.
where $J_1(x)$ is the first order Bessel function of the first kind. Now we come to a subtle point about Fourier transforms. The convolution theorem says that the Fourier transform of the product of two functions is the convolution of their transforms. The proof of the convolution theorem can be found here. This holds if the two functions have the same variable for their arguments, $\mathcal{F}\{f_1(x)f_2(x)\}=\mathcal{F}\{f_1\}*\mathcal{F}\{f_2\}$. However, if the arguments of the functions are different, the transform of their product is just the product of their transforms, $\mathcal{F}\{f_1(x) f_2(y)\}=\mathcal{F}\{f_1(x)\}\mathcal{F}\{f_2(y)\}$.
To show this, consider the Fourier transform of a product function $f_1(x)f_2(y)f_3(z)$,
Use this simulation to determine the diffraction angle $2\theta$ for the 3 21 reflection of spinel using Cu K$\alpha$ radiation. You will need to specify an $hkl$ plane where $\vec{G}_{hkl}$ is perpendicular to $\vec{G}_{3\,\overline{2}\,\overline{1}}$, otherwise the point $3\,\overline{2}\,\overline{1}$ won't appear (hint: 3 - 2 - 1 = 0). Rotate the crystal to get the 3 21 reflection on the Ewald sphere and read off the angle. The choice of the $hkl$ plane is not unique. Show that you can get the same angle for another choice of $\vec{G}_{hkl}$.
The table to the right shows the intensity of diffraction peaks that were measured in an x-ray diffraction experiment on a single crystal. The three components of the scattering vector $\Delta k$ are given in units of [1/m]. The intensities have been normalized to their largest value.
(a) What are the primitive lattice vectors of this crystal in real space?
(b) What Bravais lattice does this crystal have?
(c) How can you estimate the number of atoms in the basis? How many atoms do you estimate are in the basis of this crystal?
(d) How could you determine what atoms are in the basis and how they are arranged?
(e) Knowing how the atoms are arranged in the basis, how could you determine the point group of this crystal?